

Using Gene Expression Profiling, İdentify Genetic Changes İn Multiple Sclerosis (MS) And Determine Molecular Pathways
Clinical Medicine And Health Research Journal,
Vol. 3 No. 6 (2023),
2 December 2023
,
Page 703-708
https://doi.org/10.18535/cmhrj.v3i6.286
Abstract
Aim: In this study, an open-access dataset of RNAseq data from proggressive multiple sclerosis (MS) samples and matched normal-appearing white matter (NAWM) samples was used to identify genetic alterations that play a role in the development of MS, the prevalence is on the rise in both industrialized and emerging nations.
Material and methods: The dataset utilized in this investigation comprises nine samples of lesions and corresponding samples of NAWM tissue. The gene expression analysis was conducted on the dataset using the limma package in the R programming language. The clusterProfiler software was utilized to do enrichment analysis.
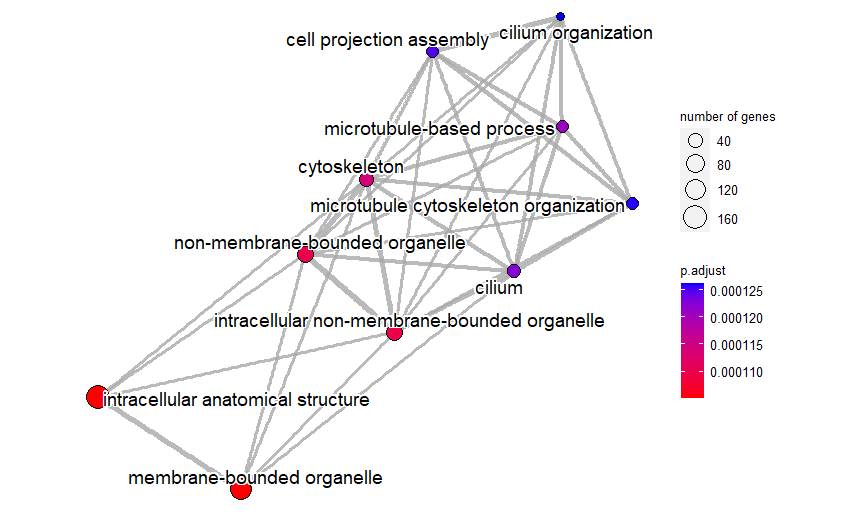
Results: 493 genes showed different up- or down-regulation in lesion tissues compared to NAWM tissues. According to the results of the enrichment analysis with these genes, the processes that play a role in progressive MS cancer are intracelluler anatomical structure, membrane bounded organelle, non-membrane bounded organelle, intracelluler non-membrane bounded organelle.
Conclusion: As a result, it can be said that the genetic structure of progressive MS tissues (lesion) changes compared to NAWM tissues, with the information obtained from gene expression analysis and enrichment analysis. It might be argued that the therapeutic efficacy of the disease can be enhanced by the comprehensive and diverse analysis of biomarkers.
- Multiple sclerosis (MS), genetic alteration, gene expression analysis, bioinformatics
How to Cite
Download Citation
References
- Article Viewed: 0 Total Download
##plugins.themes.ojsPlusA.frontend.article.downloadstatastics##
Cover Image
