Prevalence of Thalassemia in Nigeria: Pathophysiology and Clinical Manifestations
Abstract
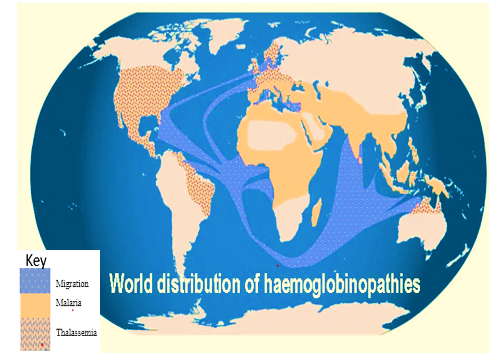
There is evidence linking genes for thalassaemia, sickle cell diseases, and glucose-6-phosphate dehydrogenase (G6PD) deficiency to a high prevalence of malaria infection. Haemoglobinopathies are hereditary conditions that mostly results in thalassaemia and sickle-cell anaemia. The current global haemoglobin gene carrier population (i.e., healthy individuals who have acquired only one mutant gene from one parent) is between 1 and 5%. Some haemoglobinopathy genes (alpha-thal, beta-thal and HbS) cause alpha-thalassaemia, beta-thalassaemia and sickle-cell anaemia, respectively. Nigerians have a prevalence of 25–30% for sickle cell anaemia (SCA), G6PD, but both alpha thalassaemia and beta thalassaemia are at the lower limit. Thalassaemia and SCA have comparable clinical manifestations. which is quite prevalent in Nigeria? This could lead to underdiagnosis of thalassaemia, which accompany hypochromia and microcytosis, that could be mistaken for iron deficiency anaemia. Depending on the levels of foetal haemoglobin and haemoglobin A2, thalassemia, iron deficiency anaemia, and sickle cell disease continue to be the most common chronic types of anaemia. This review provides details information on the prevalence of thalassaemia in Nigeria and molecular mechanisms in the expression of thalassaemia genes. The authors also suggest various possible way to minimize the occurrences of thalassaemia in Nigeria
Keywords:
- Thalassaemia, prevalence, pathophysiology, clinical manifestations and therapeutic complications.
References
Ademowo OG. (2002) Falusi AG. Erythrocyte G6PD mutations' molecular epidemiology and activity in a homogeneous Nigerian population East Afr Med J; 79:42-44
Akbari MT, Hamid M. (2012) Identification of 훼-globin chain variants: a report from Iran. Arch Iran Med; 15:564–567.
Angastinotis M. Galanello R, Eleftheriou A., (2025) Epidemiology In: Traeger-Synodinos J et al eds.Prevention of Thalassaemias and other haemoglobin disorders. Vol I. Nicosia, Cyprus:Thalassaemia International Federation Publication; :10-13
Arya V, Kumar R, Yadav RS, Dabadghao P, Agarwal S. (2009). Rare haemoglobin variant Hb I Philadelphia in north Indian family. Ann Hematol. ;88(9):927–929.
Bauer DE, Kamran SC, Orkin SH. (2012) Reawakening fetal haemoglobin: prospects for new therapies for beta-globin disorders. Blood.;120(15):2945–2953.
Borgna-Pignatti C. (2017) The current management of intermediate thalassemia. Br J Haematol 2007; 138:291- 304
Borgna-Pignatti C. (2010). The quality of life for thalassaemia major patients. Haematologica 2010; 95:345-
Bozdogan ST, Yuregir OO, Buyukkurt N, Aslan H, Ozdemir ZC, Gambin T. (2015) Alpha-thalassemia mutations in Adana province, southern Turkey: genotype-phenotype correlation. Indian J Hematol Blood Transfus.; 31:223–228.
Chen FE, Ooi C, Ha SY, Cheung BM, Todd D, Liang R, Chan TK, Chan V. (2000;) Genetic and clinical features of haemoglobin H disease in Chinese patients. N Engl J Med. 343:544–550.
Cürük MA. Hb H (2007) (beta4) disease in Cukurova, Southern Turkey. Haemoglobin.;31(2):265–71. 10.1080/03630260701297279.
Encyclopedia, M. (2021) Thalassemia: MedlinePlus Medical Encyclopedia, Medlineplus.gov. Available at: https://medlineplus.gov/ency/article/000587.htm (Accessed: 23 December 2023).
Fucharoen S, Viprakasit V. (2009) Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. (1):26–34. 10.1182/asheducation-2009.1.26
Galanello R, Origa R. (2015) Beta thalassaemia. Orphanet J Rare Dis.5:11-21
Galanello R, Pira S, Barella S et al (2010). Cholelithiasis and Gilbert’s syndrome in homozygous beta thalassaemia. Br J Haematol 2001; 115:926-928
Glickstein H, El RB, Link G et al. (2016) Action of chelators in iron loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood; 108:3195- 3203
Haidar R, Musallam KM, Taher AT. (2011) Bone disease and skeletal complications in patients with thalassaemia major. Bone; 48:425-432.
Harteveld, C. L., Higgs, D. R. (2010). α-thalassaemia. Orphanet J Rare Dis 5, 13 https://doi.org/10.1186/1750-1172-5-13.
Higgs, D. R. and Weatherall, D. J. (2009). The alpha thalassaemias. Cell Mol Life Sci, 66: 1154-1162. 10.1007/s00018-008-8529-9.
Jane BR, Noel M, Lisa AU, Michael LC, Steven AW, Peter VM. (2015) Campbell Biology. Australian and New Zealand: Pearson Higher Education.AU. Copyright; p. 1521.
Kalle Kwaifa, I., Lai, M.I. & Md Noor, S. (2020). Non-deletional alpha thalassaemia: a review. Orphanet J Rare Dis 15, 166. https://doi.org/10.1186/s13023-020-01429-1.
Karakas Z, Koç B, Temurhan S, Elgün T, Karaman S, Asker G, et al. (2015). Hipokromik Mikrositer Anemili Olgularda Alfa Talasemi Mutasyonlarının Değerlendirmesi: İstanbul Perspektifi. Turkish J Hematol. ;32(4):344–350.
Kirk P, Roughton M, Porter JB et al. (2019) Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassaemia major. Circulation; 120:1961-1968.
Kotila TR, Adeyemo AA, Mewoyeka OO, Shokunbi WA. (2019). Beta Thalassaemia trait in Western Nigeria. Afr. Health Sci;9 :46-49
Kotila TR. (2017) When the inheritance of two heterozygote states become a diagnostic problem: Misdiagnosis of the sickle cell trait. Nigeria Jornal Medical.;16 No2: 173-176
Kotila T. R. (2013). Beta thalassaemia in Nigeria: myth or fact? African journal of medicine and medical sciences, 42(4), 355–358.
Kotila TR. (2010). Guidelines for the diagnosis of the haemoglobinopathies in Nigeria. Ann Ibd Post Med. :25-29
Kwiatkowski, DP. (2005). How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet; 77:171-192
La Nasa G, Argiolu F, Giardini C et al. (2015) Unrelated bone marrow transplantation for beta thalassaemia patients: the experience of the Italian bone marrow Transplant group. Ann NY Acad Sci; 1054:186-195
Lafferty JD, Crowther MA, Ali MA., (1994) The evaluation of various mathematical RBC indices and their efficacy in discriminating between thalassaemic and non-thalassaemic microcytosis.Am J Clin Pathol 1996;106:201-205.
Masuda T, Wang X, Maeda M, et al. (2016) Transcription factors LRF and BCL11A independently repress expression of fetal haemoglobin. Science.; 351(6270):285–289.
Mettananda S, Gibbons RJ, Higgs DR. (2015) Α-globin as a molecular target in thetreatment of Β-thalassemia. Blood.;125(24):3694–701.
Mettananda S, Higgs DR. (2018) Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am.;32(2):177– 91. Available from. https://doi.org/10.1016/j.hoc.2017.11.003.
Nainggolan IM, Harahap A, Ambarwati DD, et al. (2013) Interaction of Hb Adana (HBA2: c.179G>A) with deletional and nondeletional (+)- thalassemia mutations: diverse haematological and clinical features. Haemoglobin. ; 37:297–305.
Novartis. (2021). Available at: https://www.novartis.com/ (Accessed: 8 September 2023).
Omotade OO. (1998) Kayode CM, Falade SL, Ikpeme S, Adeyemo AA, Akinkugbe FM. Routine screening for sickle cell haemoglobinopathy by electrophoresis in an infant welfare clinic. West AfrJ Med.; 17:91-94
Rathod DA. (2017). Amarjeet K, Patel V et al. Usefulness of cell counter-based parameters and formulas in detection of thalassaemia trait in areas of high prevalence. Americal Jornal Clinical Pathology. 128:585-589
Schrier, S. (2002) "Pathophysiology of thalassemia", Current Opinion in Hematology, 9(2), pp. 123-126. doi: 10.1097/00062752-200203000-00007
Singer ST. (2009) Variable clinical phenotypes of α-thalassemia syndromes. ScientificWorldJournal. ;9(March):615–625.
Singh SA, Sarangi S, Appiah-Kubi A, Hsu P, Smith WB, Gallagher PG, Chui DHK. (2018) Hb Adana (HBA2 or HBA1: c.179G > A) and alpha thalassemia: Genotype-phenotype correlation. Pediatr Blood Cancer. ;65(9):e27220. doi: 10.1002/pbc.27220.
Smith EC, Orkin SH. (2016). Haemoglobin genetics: recent contributions of GWAS and gene editing. Hum Mol Genet. 2016;25(R2): R99–105.
Taher A, Isma’eel H, Mehio G et al. (2016) Prevalence of thromboembolic events among 8860 patients with thalassaemia major and intermedia in the Mediterranean and Iran. Thrombosis and Haemostasis 488-491
Tan JA, Kho SL, Ngim CF, Chua KH, Goh AS, Yeoh SL, George E. (2016) DNA studies are necessary for accurate patient diagnosis in compound heterozygosity for Hb Adana (HBA2:c.179 > A) with deletional or nondeletional alpha-thalassaemia. Sci Rep. ;6:26994.
Thalassemia - Symptoms and causes (2021). Available at: https://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/syc-20354995 (Accessed: 23 December 2023).
Thalassemias | NHLBI, NIH (2014). Available at: https://www.nhlbi.nih.gov/health-topics/thalassemias (Accessed: 18 December 2023).
Toumba M, Skordis N. (2010). Osteoporosis syndrome in thalassaemia major: An overview. J Osteoporos; 201:537-544.
Vernimmen D. Globins, (2018) from genes to physiology and diseases. Blood Cells Mol Dis.70:1. Available from. https://doi.org/10.1016/j.bcmd.2017.02.002.
Viprakasit V, Tanphaichitr VS, Chinchang W, Sangkla P, Weiss MJ, Higgs DR (2004). Evaluation of alpha haemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with β thalassemia. Blood. ;103(9):3296–3299
Voskaridou E, Terpos E. (2014) New insight into the pathophysiology and management of osteoporosis in patients with thalassaemia. Br Jhaematol ;127:127-139.
Weatherall DJ, Clegg JB. (2011). The thalassaemia syndrome. 2001; 4th edn. Blacwell Scientific Publications, Oxford.
Yatim NF, Rahim MA, Menon K, et al. (2014) Molecular characterization of 훼- and 훽-thalassaemia among Malay patients. Int J Mol Sci; 15:8835–8845.
Zhou D, Liu K, Sun CW, et al. (2010) KLF1 regulates BCL11A expression and gamma- to beta globin gene switching. Nat Genet.;42(9):742–744.
-
Article Viewed: 0
Total Download
##plugins.themes.ojsPlusA.frontend.article.downloadstatastics##